
Research
Research in the Carter group currently focuses entirely on using quantum-based simulations methods to discover and design materials for sustainable production of fuels and chemicals through photo/electro/thermo-catalysis, as well as carbon dioxide mineralization.
The work builds on Carter’s pioneering development of efficient and accurate first principles quantum mechanics techniques for electron correlation, embedded correlated wavefunction, and orbital‐free density functional theories.
Open-sourced codes developed by the Carter group are available from github: https://github.com/EACcodes
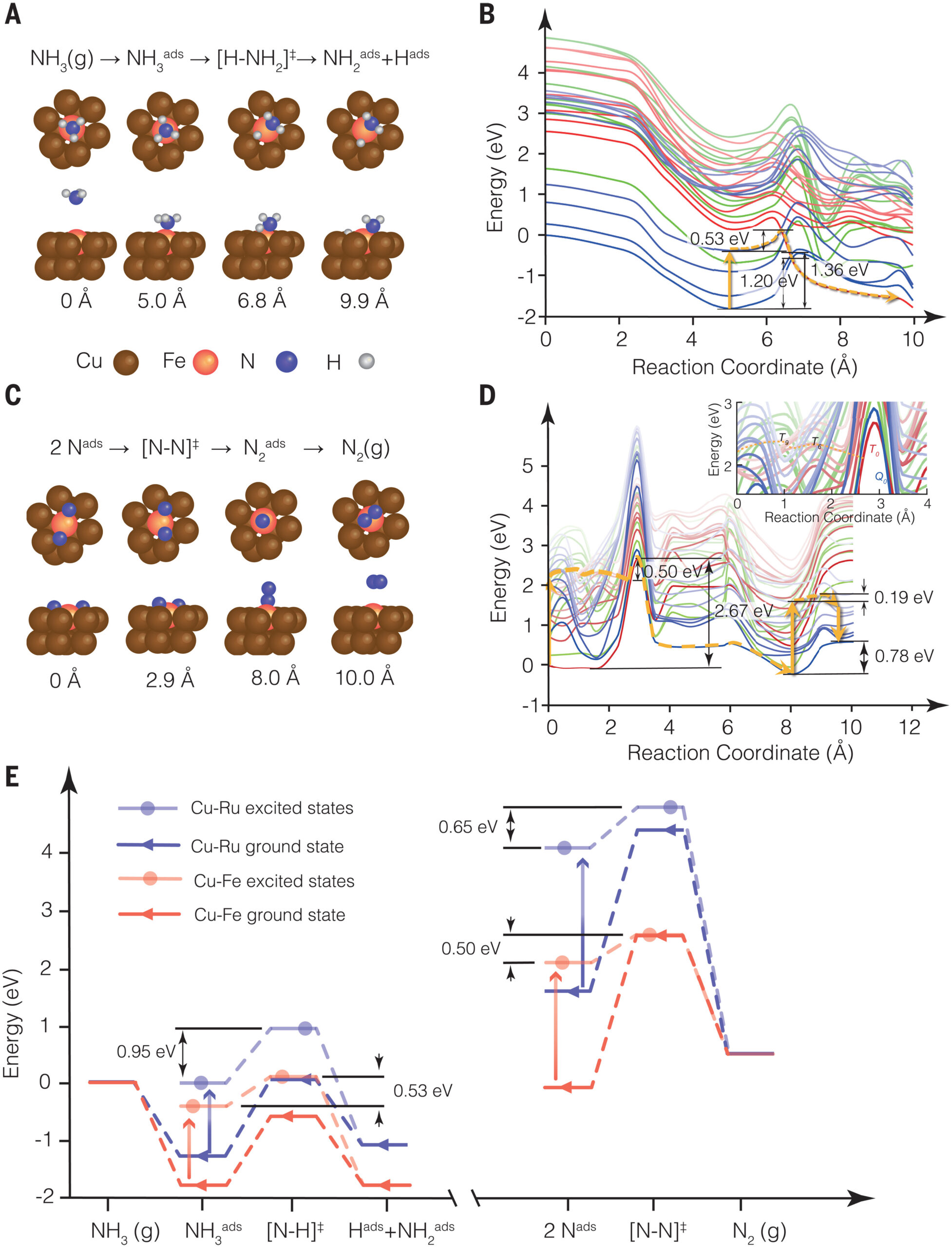
Y. Yuan, L. Zhou, J. L. Bao, J. Zhou, A. Bayles, L. Yuan, M. Lou, M. Lou, S. Khatiwada, H. Robatjazi, E. A. Carter, P. Nordlander, and N. J. Halas, “Earth-abundant photocatalyst for H2 generation from NH3 with light-emitting diode illumination,” Science, 378, 889 (2022). doi: 10.1126/science.abn5636
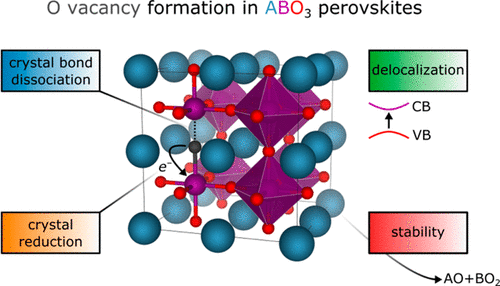
R. B. Wexler, G. S. Gautam, E. B. Stechel, and E. A. Carter, “Factors Governing Oxygen Vacancy Formation in Oxide Perovskites,” J. Am. Chem. Soc., 143, 13212 (2021). doi: 10.1021/jacs.1c05570
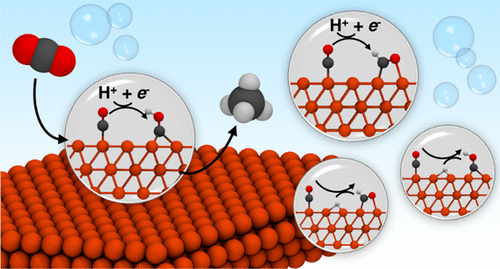
Q. Zhao, J. M. P. Martirez, and E. A. Carter, “Revisiting Understanding of Electrochemical CO2 Reduction on Cu(111): Competing Proton-Coupled Electron Transfer Reaction Mechanisms Revealed by Embedded Correlated Wavefunction Theory,” J. Am. Chem. Soc., 143, 6152 (2021). doi: 10.1021/jacs.1c00880
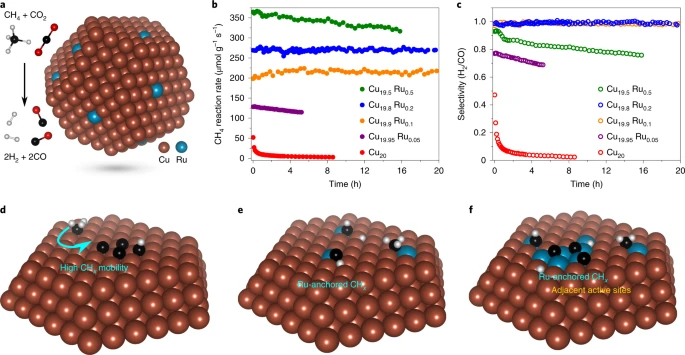
L. Zhou, J. M. P. Martirez, J. Finzel, C. Zhang, D. F. Swearer, S. Tian, H. Robatjazi, M. Lou, L. Dong, L. Henderson, P. Christopher, E. A. Carter, P. Nordlander, and N. J. Halas, “Light-driven methane dry reforming with single atomic site antenna-reactor plasmonic photocatalysts,” Nat. Energy., 5, 61 (2020). doi: 10.1038/s41560-019-0517-9
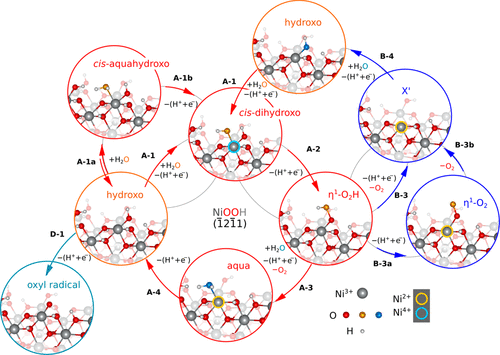
J. M. P. Martirez and E. A. Carter, “Unraveling Oxygen Evolution on Iron-Doped β-Nickel Oxyhydroxide: The Key Role of Highly Active Molecular-like Sites,” J. Am. Chem. Soc, 141, 693 (2019). doi: 10.1021/jacs.8b12386